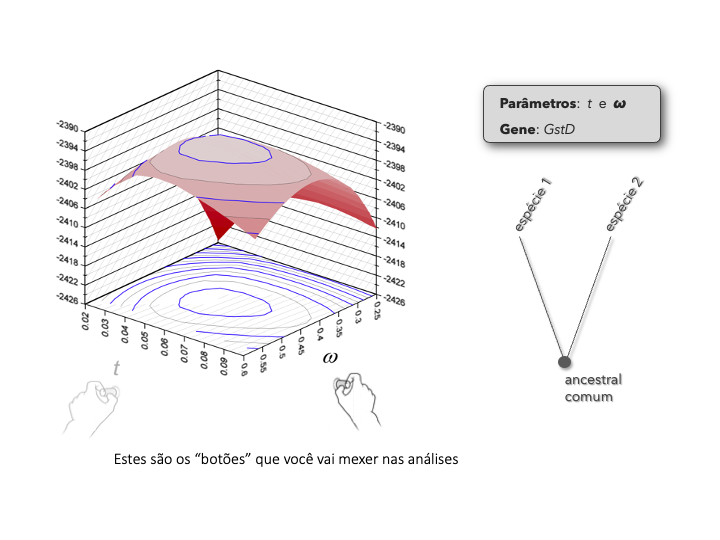
O objetivo desta atividade é te ajudar a entender como utilizar diferentes modelos de códon, e como realizar testes de seleção natural usando o programa codeml do PAML. Esta atividade também foi planejada para desenvolver habilidades analíticas gerais, que são relevantes para realizar análises com o PAML, assim como outros pacotes de software similares (ex: Hyphy).
Este tutorial é dividido em 4 exercícios:
A próxima seção, Acessando os arquivos, descreve como acessar e trabalhar com os arquivos necessários para cada exercício. Você irá acessar os arquivos de forma diferente, dependendo de onde estiver fazendo os exercícios (nos laboratórios do workshop ou em casa).
Importante! Para rodar o codeml ao longo dos exercícios, você precisará modificar arquivos input e criar arquivos output.
Para isso, recomendamos que você:
Recursos adicionais
Slides para a atividade de aprendizado do PAML: 2024_slides
Cópia do capítulo que acompanha estes exercícios: Book_Chapter.pdf
Página com documentos úteis e vários outros recursos adicionais.
1. Se você estiver fazendo esta atividade NO WORKSHOP: as máquinas virtuais que vamos usar no workshop de 2022 possuem um link simbólico no diretório home chamado " moledata" que contém os arquivos do curso. Nele você encontrará diretórios para os diversos laboratórios deste workshop (e.g., MSAlab, revbayes, PamlLab, etc.).
Para visualizar a lista de laboratórios digite o seguinte comando:
ls ~/moledata
Para visualizar os conteúdos do laboratório Paml, digite:
ls -1 ~/moledata/PamlLab
O comando acima irá listar os diretórios para cada exercício:
ex1
ex2
ex3
ex4
Estes arquivos já estão na máquina virtual que você está usando. Entretanto, recomendamos que você faça cada exercício em um diretório separado que você irá criar. Você pode escolher o nome do diretório, mas procure escolher algo informativo (ex: ~/PAML_ex1).
Para copiar os arquivos necessários para o exercício 1 digite:
cp ~/moledata/PamlLab/ex1/* ~/PAML_ex1
Agora você está pronto(a/e) para fazer o exercício 1 dentro da pasta ~/PAML_ex1.
2. Se você estiver fazendo esta atividade INDEPENDENTEMENTE do workshop: você pode baixar os diretórios de todos os exercícios aqui, ou os arquivos individuais para cada exercício aqui.
De qualquer forma, ainda recomendamos que você faça cada exercício em um diretório de trabalho separado que você irá criar (ex: PAML_ex1), e trabalhe com cópias dos arquivos necessários dentro desse diretório.
O objetivo deste exercício é usar o CODEML para avaliar o likelihood de sequências do gene GstD1 com diversos valores ω. Faça um gráfico dos valores de log-likelihood contra os valores de ω e determine a estimativa de ω com likelihood máximo. Verifique o seu resultado rodando o algoritmo de hill-climbing do CODEML.
Localize os arquivos de input para o Exerício 1: ex1_codeml.ctl, e seqfile.txt e familiarize-se com eles. Preste muita atenção no conteúdo do arquivo controle: ex1_codeml.ctl.
Lembre-se de criar um diretório para armazenar os resultados, e coloque todos os seus arquivos nele. Agora abra uma janela do terminal, e vá até o diretório que contém os seus arquivos. Quando você estiver pronto(a/e) para rodar o CODEML, delete o prefixo ex1 do arquivo controle, para que o nome seja apenas codeml.ctl. Agora você pode rodar o CODEML.
Familiarize-se com os resultados, que estarão no arquivo results.txt (a menos que você tenha editado o arquivo controle para que os resultados sejam registrados em outro arquivo) - veja o Arquivo de ajuda – Atividade 1) para mais detalhes. Identifique a linha dentro do arquivo de resultados que dá o valor de likelihood para o dataset do exemplo.
Agora mude e salve o arquivo controle, e rode o CODEML novamente usando outro valor fixo de ω (o guia rápido) para o arquivo controle pode ser útil aqui). O objetivo é computar o likelihood do dataset dado um valor fixo de ω. Mude o arquivo controle da seguinte forma:
outfile =) no arquivo controle, senão você irá sobrescrever os resultados antigos e perdê-los!omega = no arquivo controle. Os valores para este exercício estão nos comentários no final do arquivo controle ex1_codeml.ctl. Repita o passo 4 para cada valor de ω de acordo com os comentários no arquivo controle (ex: ω = 0.005, 0.01, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 2.0).
Use seu programa de planilhas ou pacote de análises estatísticas favorite para plotar os valores de likelihood (eixo y) contra os valores fixos de omega (eixo x). Use uma escala logarítmica para o eixo x (mas não transforme os valores de ω). Seu gráfico deverá ficar mais ou menos assim: ex1 plot template.pdf (obs: os pontos do gráfico foram omitidos intencionalmente desse arquivo, você precisa gerá-los no seu próprio gráfico).
A partir do seu gráfico, responda a seguinte questão:
Agora mude o arquivo controle para que o CODEML use seu algoritmo de hill-climbing para encontrar o MLE; especificando fix_omega = 0 no arquivo controle. Compare o resultado com a sua resposta do passo 7.
Neste exercício você irá investigar a sensibilidade do ω em relação à proporção de transições/transversões (kappa), e às frequências de códon assumidas (π's). Após coletar os dados necessários, determine quais pressupostos resultam nos maiores e menores valores de S, e qual efeito isso tem no valor de ω.
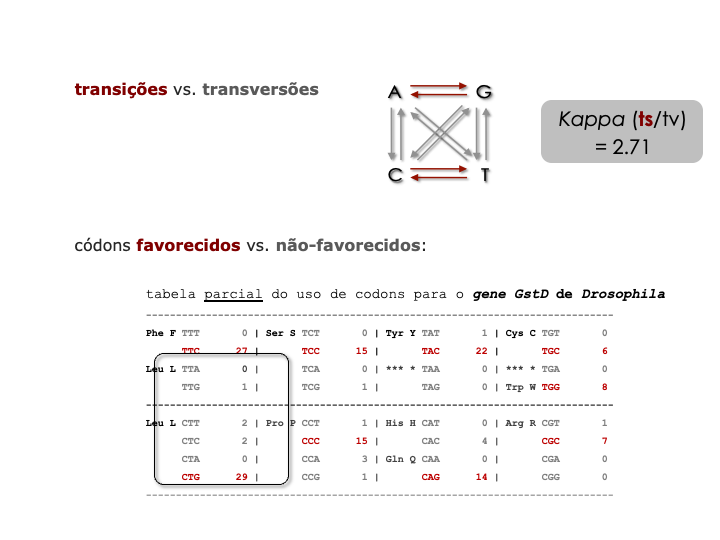
Encontre os arquivos para o Exercício 2 (ex2_codeml.ctl, seqfile.txt) e familiarize-se com eles. Recomendamos que você crie um novo diretório para o Exercício 2. Quando você estiver pronto(a/e) para rodar o CODEML, delete o prefixo ex2_ para que o nome do arquivo controle seja codeml.ctl.
Rode o CODEML usando as configurações no arquivo controle para o Exerício 2. Familiarize-se com os resultados, usando o Arquivo de ajuda – Atividade 2). Além de localizar o valor de likelihood, você deve identificar a parte do arquivo de resultado que apresenta estimativas para os seguintes valores:
número de sítios sinônimos e não sinônimos (S e N)
taxas de substituições sinônimas e não sinônimas (dS e dN)
Como no exercício 1, você precisará modificar e salvar os arquivos controles e rodar novamente o CODEML. O "guia rápido" para o arquivo controle pode te ajudar aqui! O objetivo é calcular o likelihood do dataset sob os pressupostos de diferentes modelos. Para fazer isso, você deve:
mudar o nome do arquivo de resultados (usando outfile = no arquivo controle), para evitar que os novos resultados sobrescrevam os antigos
mudar os pressupostos dos modelos sobre as frequências dos codons (usando CodonFreq =) e o valor de kappa (através da opção kappa = e fix_kappa =)
Repita o passo 3 para cada conjunto de pressupostos sobre as frequências dos codons e para cada valor de kappa mostrados na figura abaixo. Você também pode encontrar esses valores nos comentários no final do arquivo controle.

Usando seu programa de planilhas favoritos, crie uma tabela como a Tabela E2 (presente neste arquivo: ex2 table template.pdf e preencha-a com os seus resultados.
Use sua tabela para determinar:
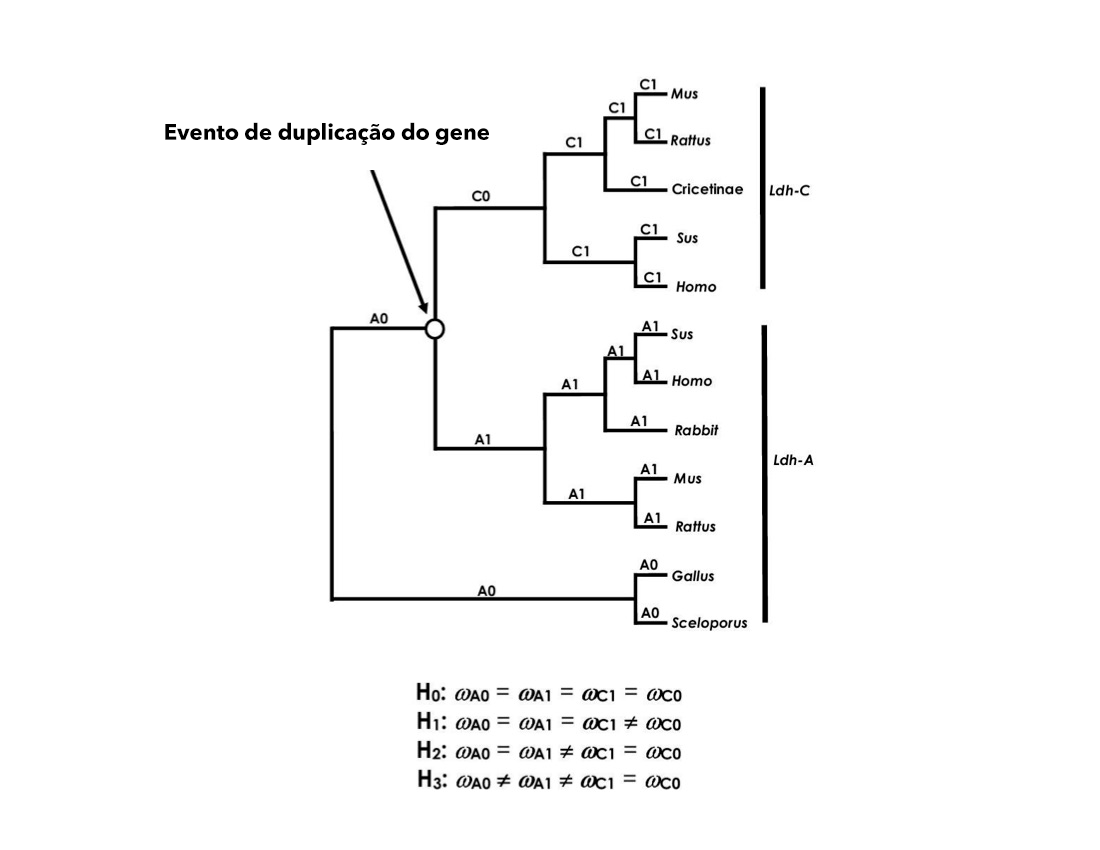
O objetivo deste exercício é usar três LRTs (Likelihood Ratio Test) para avaliar as seguintes possibilidades: (1) a taxa de mutação do gene Ldh-C aumentou em relação ao Ldh-A, (2) um surto de seleção positiva para divergência funcional ocorreu durante o evento de duplicação que deu origem a Ldh-C, e (3) houve uma mudança de longo prazo nas pressões seletivas, seguindo o evento de duplicação que deu origem a Ldh-C.
Baixe os arquivos para o Exercício 3 no website do curso (ex3_codeml.ctl, seqfile.txt, treeH0.txt, treeH1.txt, treeH2.txt, treeH3.txt). Os arquivos de árvores representam diferentes hipóteses, denominadas H0, H1, H2 e H3 (veja o diagrama abaixo, ou faça o download do arquivo genetree. Os conceitos evolutivos descritos acima são cobertos por essas quatro hipóteses:
H0: pressões seletivas homogêneas ao longo da árvore (conceito = quaisquer diferenças nas taxas de substituição são devidas a mudanças nas taxas de mutação).
H1: mudanças episódicas nas pressões seletivas no gene Ldh-C (conceito = apenas no ramo que segue imediatamente o evento de duplicação gênico).
H2: mudanças de longo prazo nas pressões seletivas apenas no gene Ldh-C (conceito = Ldh-C está sob uma nova pressão seletiva comparado aos seus ancestrais, enquanto Ldh-A permanece sujeito aos níveis ancestrais de seleção).
H3: mudanças de longo prazo nas pressões seletivas em Ldh-C e Ldh-A (conceito = ambas as linhagens parálogas passaram por novas pressões seletivas, diferentes um do outro e também do ancestral).
Rode CODEML usando as configurações do arquivo controle para o Exercício 3. Familiarize-se com os resultados (ex3_HelpFile.pdf). Além de localizar o valor de likelihood, você deve identificar as estimativas de ω específicas para cada ramo (quando você rodar pela primeira vez - H0, os valores de ω serão os mesmos em todos os ramos).
Assim como nos exercícios anteriores, você precisará modificar e salvar os arquivos controles e rodar novamente o CODEML. O "guia rápido" para o arquivo controle pode te ajudar aqui! O objetivo é calcular o likelihood e estimar os parâmetros ω sob diferentes modelos que descrevem como as pressões seletivas mudam em diferentes partes da árvore. Como as informações relevantes para os modelos estão contidas nos arquivos de árvores, você precisará espeficiar cada um desses arquivos a cada análise, e mudar o arquivo conrole para que ele leia o arquivo de árvore correspondente.
outfile = no arquivo controle), para evitar que os novos resultados sobrescrevam os antigos.treefile = e model =) especificados no arquivo controle.Repita o passo 3 para cada uma das quatro árvores presentes na pasta do Exercício 3. Novamente, mantenha um registro dos seus resultados usando uma tabela como a Tabela 3 mostrada nos slides (veja ex3 table template.pdf). Além disso, faça testes de likelihood (Likelihood Ratio Tests - LRT) com as hipóteses abaixo. Veja as anotações da aula para detalhes adicionais sobre LRTs. Use 1 grau de liberdade para obter o P-valor para cada LRT (você pode usar esta calculadora de Chi-quadrado para computar os P-valores).
Use sua tabela de resultados para determinar:
Qual(is) modelo(s) são suportados pelos dados?
Qual cenário evolutivo é a melhor explicação para a evolução da família gênica Ldh?
Existe evidência de seleção positiva durante a história evolutiva da família Ldh?
Existe algum cenário no qual os genes Ldh poderiam ter evoluído por seleção positiva e que seria indetectável através de LRTs?
O objetivo deste exercício é usar uma série de LRTs para testar sítios evoluíndo por seleção positiva no gene nef. Se você encontrar evidência significativa de seleção positiva, identifique os sítios envolvidos usando os métodos empíricos de Bayes.
Obtenha todos os arquivos para o exercício 4 (ex4_codeml.ctl, seqfile.txt, treeM0.txt, treeM1.txt, treeM2.txt, treeM3.txt, treeM7.txt, treeM8.txt). Quando estiver pronto(a/e) para rodar o CODEML, lembre-se deletar o prefixo ex4_ para que o nome do arquivo controle seja codeml.ctl.
Se você estiver planejando rodar dois ou mais modelos ao mesmo tempo, crie um diretório separado para cada modelo e coloque dentro dele uma cópia do arquivo de sequência, o arquivo de árvore e o arquivo de controle.
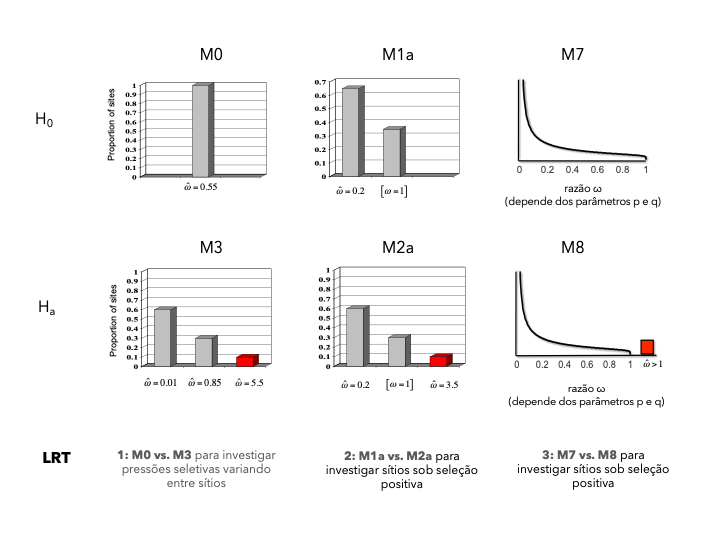
Assim como em todos os exercícios anteriores, você precisará modificar os arquivos controles e rodar novamente o CODEML várias vezes. Neste caso, você estará ajustando (testando o fit) seis modelos diferentes (M0, M1a, M2a, M3, M7 e M8) ao dataset. O "guia rápido" para o arquivo controle pode te ajudar aqui! ("guia rápido 2")
Se você estiver rodando suas análises sequencialmente no mesmo diretório, você deve mudar o nome do arquivo de resultados (através da opção outfile = no arquivo controle), para evitar que os novos resultados sobrescrevam os antigos.
Especifique o arquivo de árvore utilizando a opção treefile = do arquivo controle. As árvores necessárias para cada modelo, com seus respectivos comprimentos de ramo, estão disponíveis no diretório do Exercício 4 para você usar (para cada modelo, especifique uma das árvores disponíveis). Isso irá tornar sua análise muito mais rápida, e quem sabe você consegue tomar aquela cervejinha no fim do dia :) Veja o arquivo controle para mais detalhes sobre os nomes dos arquivos de árvore.
Especifique o modelo de códon para cada análise usando a opção NSsites =.
Deixe o valor de kappa fixo na estimativa de maximum likelihood, usando kappa =. Novamente, isso irá tornar a análise mais rápida. Veja o arquivo controle para saber quais valores de kappa você irá colocar para cada modelo.
Para alguns modelos, você também precisará especificar o número de categorias (ncatG) na distribuição ω:
para M3: ncat = 3
para M7: ncatG = 10
para M8: ncatG = 10
Quando a análise estiver completa, mude o nome do arquivo rst, para evitar sobrescrever o seu conteúdo durante as análises consecutivas.
Repita os passos para cada um dos seis modelos de códon listados acima.
Mantenha um registro dos seus resultados (Arquivo de ajuda - Atividade 4) usando uma tabela como a Tabela E4 mostrada nos slides (veja a tabela ex4 table template.pdf).
Além disso, faça os seguintes testes de likelihood (LRT):
M0 vs. M3 (4 graus de liberdade)
M1a vs. M2a (2 graus de liberdade)
M7 vs. M8 (2 graus de liberdade)
Abra o arquivo rst gerado durante o modelo M3 (Arquivo de ajuda - rst). Localize as colunas com as probabilidades posteriores de cada sítio sob as três categorias de sítios deste modelo. Use esses dados para produzir um gráfico para o gene nef, como mostrado abaixo (obs: seu gráfico vai ser semelhante, mas não idêntico ao mostrado abaixo).
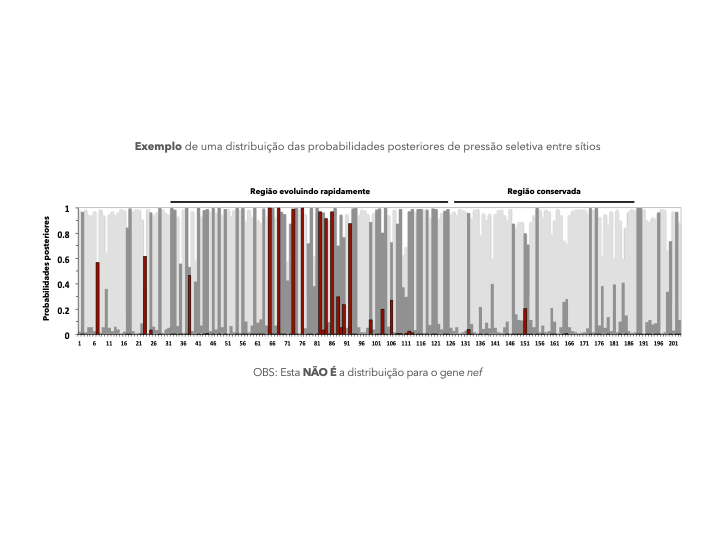
Por último, tente fazer uma síntese de todos os seus resultados e busque uma interpretação biológica que poderia ser publicada, por exemplo, em um artigo científico. Inspire-se nas duas questões gerais abaixo, e, se possível, converse com outros estudantes do workshop (ou da sua faculdade/laboratório etc) para construir ideias juntos(as/es). Afinal, a ciência é um lugar de colaboração!
Que conclusões biológicas são bem suportadas por esses dados?
Quais aspectos dos resultados você consegue interpretar de acordo com seu conhecimento biológico desse sistema ou de sistemas similares?
Agora que você possui um pouco de experiência com modelos de códons, você está pronto(a/e) para tentar um tutorial cobrindo tópicos mais avançados. O tutorial avançado foca em detectar evolução adaptativa episódica utilizando o modelo "Branch-Site A".
O tutorial também inclui atividades adicionais para:
identificar e marcar ramos em árvores filogenéticas usadas como input para modelos de códon branch-site
detectar instabilidades nas suas estimativas de parâmetros
realizar análises de robustez
usar o método SBA (smoothed bootstrap aggregation) para corrigir suas análises de acordo com incertezas na estimativa de parâmetros e instabilidade nos modelos de códon
Os protocolos para cada atividade estão publicados na revista Protocols in Bioinformatics (UNIT 6.15). Essa publicação também possui recomendações de "boas práticas" ao realizar análises evolutivas de larga escala para evolução episódica adaptativa usando PAML.
Veja o PDF para Protocols in Bioinformatics (UNIT 6.15) aqui: UNIT 6.15
Os arquivos necessários para o tutorial avançado estão disponíveis neste repositório Bitbucket: link-repositório