Workshop on Molecualr Evolution at the MBL
Follow this link to go to back to PAML lab.
Follow this link to go to back to workshop schedule page.
Follow this link to go to back to the Bielawski faculty page.
Follow this link to go to an on-line Chi-square calculator..
Here are a couple of ways in which you might choose to plot your results for Exercise 1.
EXCEL:
Input your data:
`Column A --> omega values`
`Column B --> lnL scores`Select both columns of data.
Use drop down menus to select the chart type:
`Insert --> Charts --> Scatter (choose lines)`Specify a log scale for the x-axis:
` Right click on X-axis (omegas) --> Format Axis --> Logarithmic scale`R:
`x_vector <- c(your parameter values, separated by commas)`For example: x_vector <- c(.005,.006,.007,.008)
`y_vector <- c(your likelihoods, separated by commas)`For example: y_vector <- c(.005,.006,.007,.008)
`plot(x = vector, y =y_vector, log="x", type="b")`Labelling the branches of a Newick-formatted tree-file for use with a “branch model” or a “branch-site codon model" is tricky. It becomes very challenging when the tree is very large.
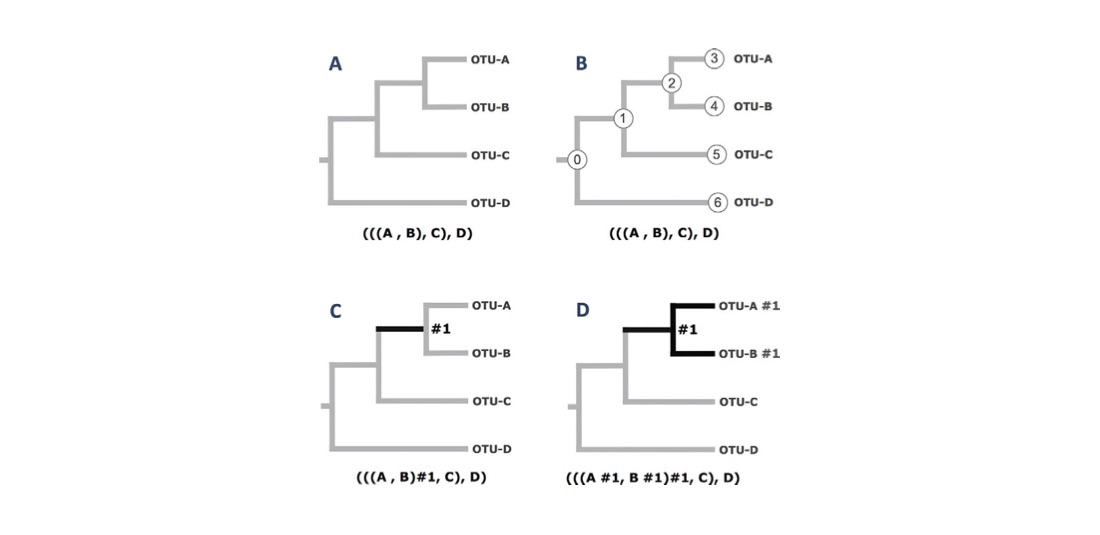
Recall that the objective of Exercise 3 is to illustrate how a “branch model” can be used to specify specific biological hypotheses that can be formally tested via a Likelihood Ratio Test (LRT). The figure below illustrates how “branch marks” within a three file are used to denote specific branches of the tree. Such “branch marks” are required by the “branch models” or a “branch-site codon models"
In the figure above, the operational taxonomic units (OTUs) at the tips of the tree are labeled A, B, C, and D. Those branches where a user wants to specify unique model parameters are referred to a “foreground” (FG) branches. The FG branches of each phylogeny are shown in bold. The Newick tree (parenthetical tree notation) corresponding to each phylogeny is shown below each case. In this example, the foreground branch is indicated within a Newick tree using the #1 label (aka “a branch mark”). (A) An unlabeled phylogenetic tree. This tree will have no branch-specific parameters. (B) The same (unlabeled) tree as in A, but with node IDs shown for both tip and internal nodes. (C) A phylogenetic tree having a single FG branch, shown in bold. The FG branch will have an independent ω in parameter in a “branch model”. The user must supply the label for the FG branch in the Newick tree. In this case the label is “#1”. Note that the FG branch in this tree is equivalent to branch 2 in tree B above. (D) A phylogenetic tree having a set of three FG branches, shown in bold. All three foreground branches will have the same ω in parameter in a “branch model” because they all have the same label (i.e., they all have #1).
Nick Myerson (a former workshop TA) wrote a guide to labelling tree files that is very helpful. You can download that document here.
Additional information about labelling trees (or entire clades) can be found in the PAML manual (pamlDOC.pdf). For larger trees, the assistance of software might be necessary. One option is the program called “DendroCypher” (repository-link).
This is not part of the exercises because appropriately formatted sequence data files were provided. But this information will be helpful when it is time to run these kinds of analyses on your own datasets.
To fit codon models in PAML, you need a codon alignment that is “in-frame” (i.e., a nucleotide alignment that, when translated, corresponds perfectly to amino acid sequences with no frame-shifts or in-fame stop codons). You can't just use a nucleotide alignment because most alignment programs do not keep track of the reading frame. Such DNA alignments will, in almost all cases, insert gaps the will disrupt the reading frame.
One way to obtain a properly aligned codon sequence is to translate each sequence to amino acids (e.g., using SeaView) and first align the amino acid sequences (e.g., via MAFFT or Muscle). Once you have an amino acid alignment, the second step is to use software to force each DNA sequence into an alignment that matches the amino acid alignment. There are several options for this; one popular method is the Pal2Nal server.
For Pal2Nal, you will upload your protein alignment and nucleotide sequences to the server. Obviously, these nucleotide sequences must be multiples of three, contain no stop codons, correspond exactly to one of the amino acid sequences, and have the same name as the corresponding amino acid sequence. Pal2Nal will then produce a codon-aware DNA alignment that corresponds to the amino acid alignment that as uploaded. The resulting alignment will be suitable for input to PAML (if appropriately formatted).
You can download the necessary files from an archive
You can download individual files here.
The PAML lab slides: pamlDEMO.pdf.
Here is a copy of the book chapter that accompanies the lab exercises: Book_Chapter.pdf.
Here is a copy of the PAML user manual: pamlDOC.pdf.
Here is a copy of the PAML FAQ: pamlFAQs.pdf.
Here is a copy of the paper for PAML4: Yang2007MBE.
A book chapter including a discussion of workflow, experimental design and best practices: UNIT6.15.pdf.
An example of a 2-phase approach to "best practices" focused on quality control and robustness: BakerGenetics2016.
A comprehensive review of the major inference challenges under codon models: Jones et.al. 2019.
An “advanced" codon model tutorial is available via this Bitbucket repository: repository-link
The program DendroCypher is available for download here: repository-link
My home page: www.bielawski.info.
Ziheng Yang's page: https://profiles.ucl.ac.uk/7354-ziheng-yang-frs.
PAML page: https://github.com/abacus-gene/paml.
HyPhy page: http//hyphy.org.
DataMonkey Adaptive Evolution Server: www.datamonkey.org.
COLD program documentation and download: www.mathstat.dal.ca/~tkenney/Cold/.
The PAML Discussion Group (Google group): https://groups.google.com/forum/#!forum/pamlsoftware.